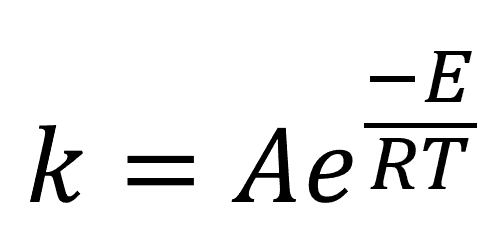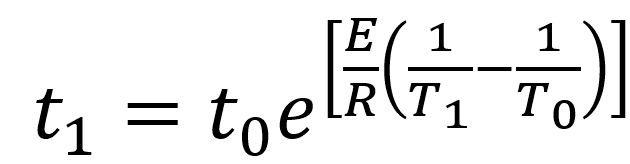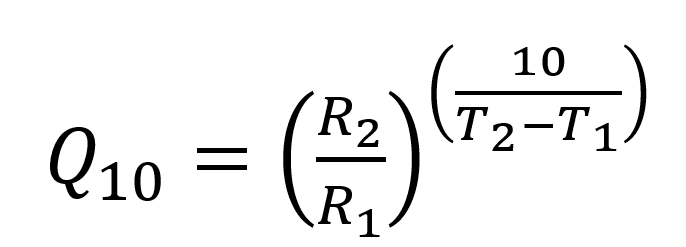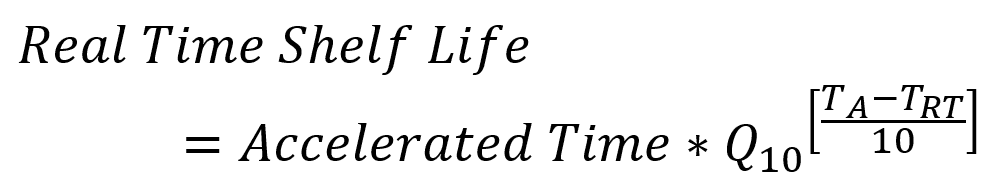Posted by Dr. Gavin Braithwaite on | Comments Off on Plastic on the Brain
Over the past 50 years, the prevalence of plastics in our consumer waste stream has increased significantly as these materials have become more widely used in products, packaging, and single-use disposable items like water bottles. There has been considerable discussion about the fate of plastics, with coverage often focusing on recycling facilities, landfills, and ocean “garbage patches” in places like the North Atlantic Gyre. More recently, however, attention has shifted toward microplastics as a critical component of plastic pollution, including emerging evidence that these particles can accumulate in human organs such as the liver, kidneys, and brain.
What Exactly Are Micro- and Nanoplastics?
Microplastics are polymer-based particulates with a size range between 0.1 micrometers to 5 mm; while nanoplastics extend from 0.1 micrometers to 1 nm. These particles arise from the breakdown of larger plastic items, as well as from direct sources such as fibers shed from textiles, industrial abrasives, cosmetic additives, and wear debris from polymer components.
Although currently the impact of these materials is largely unknown, toxicologists and clinical researchers are investigating the potential harm of microplastics and nanoplastics on human and animal health. Irrespective of the size of the particles, a key part of this investigation is to identify and quantify the types of polymers making up the particulates, as toxicological studies consider the amount of a particular compound in the body when evaluating the toxicological response (e.g. inflammation, necrosis, etc.), following the old adage “dosis sola facit venenum”, or “the dose makes the poison.” In other words, for a toxic compound to be harmful in the body, it has to reach a threshold in concentration in a biological system.
How Do We Look for Microplastics and Nanoplastics?
Historically, much of the work to detect microplastics in tissues has relied on manual microscopy, sometimes preceded by tissue digestion processes to allow separation of unknown particles. This approach is laborious and faces difficulties in accurately identifying and quantitating the microplastics in the tissue. These difficulties stem from sub-sampling challenges and concerns about the quality and consistency of the recovery process. Due to the resolution limits of microscopy, this approach is unable to see nanoplastics, or even the lower range of microplastics.
These smaller particles are of particular interest because a large fraction of the material may exist below conventional optical resolution limits. Although the total volume of these particles is not substantial, this population may be significant in terms of the number of particles. Capturing this population is important, because absolute dose is only part of the concern, and the surface area-to-volume ratio may also be significant. Smaller particles present more surface area per unit mass, which can influence reactivity and biological interactions. One can see that intuitively because a medical implant made from a polymer has been validated to be safe, but it is not clear whether nanoplastics of the same polymer, capable of migrating to different organs, should be considered safe.
The biological impact of micro- and nano-plastics are not yet well understood, but having analytical tools to identify and quantify them is an essential step toward determining if they contribute to clinical issues.
Py-GC/MS: Looking Inside Tissues without Seeing the Particles
Consequently, researchers are turning to other techniques to identify and quantitate these smaller particles. An emerging technique is pyrolysis gas chromatography with mass spectroscopy (Py-GC/MS), which allows analysis of particulates (both micro and nano) in their native tissue matrices. In Py-GC/MS, the sample is thermally decomposed, and the resulting fragments are separated and analyzed to generate polymer-specific fingerprints.
This approach offers several advantages for micro- and nanoplastic analysis:
Direct analysis of intact tissues or partially processed matrices.
Polymer-specific identification and quantitation, even when particles are below optical resolution.
Simultaneous detection of multiple common plastic types.
Using Py-GC/MS, it is possible to distinguish and measure polymers such as polyurethanes, polymethyl methacrylates, polystyrenes, polyvinyl chloride, polyethylene, polypropylene, nylons, polyesters, and polycarbonates in biological samples.
Cambridge Polymer Group has recently developed assays using Py-GC/MS to investigate micro and nano-plastics in lung tissue from certain high-risk industries, providing a path to more robust exposure assessment and materials for subsequent toxicological evaluation.
What Is Being Found in Human Organs?
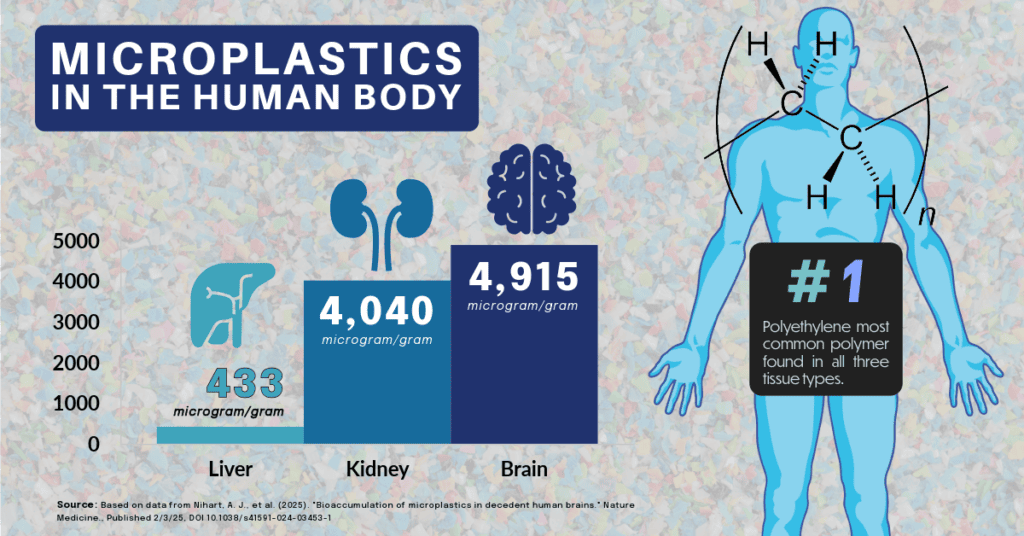
A recent study published by Nihart and colleagues (2025)[1] used Py-GC/MS to examine micro and nanoplastics in human livers, kidneys and brains. They obtained tissues from autopsy specimens from 2015-2024.
Their analysis showed consistent levels of plastic particulates in the liver and kidney specimens at concentration levels of 433 and 4,040 microgram/g, respectively. The plastic levels in the brain were much higher, reaching median levels of 4,915 microgram/g in the 2024 samples. The authors note that brain tissue from patients diagnosed with dementia had a notably higher concentration of particulates.
Interestingly, polyethylene was the most common type of polymer found in all three tissue types, particularly for the brain tissue. No hypothesis is offered by the authors as to why this polymer may be more prevalent.
Polyethylene is a commonly used commercial polymer with many applications ranging from supermarket bags to water bottles. Arguably one would expect to see more polyesters and polyvinyl chlorides, given their abundance in single-use packaging systems. One potential source of polyethylene particulates is from hip and knee replacements, where the bearing surface is often comprised of a form of polyethylene, and if this bearing surface undergoes wear, polyethylene particles are generated. The study did not examine this potential source. If these particles are indeed from orthopedic implants the data suggests that particles can migrate throughout the body, irrespective of source. A future study may attempt to correlate observed polyethylene particulates with the presence of a hip or knee replacement in the patient. As polyethylenes used in these implants have improved in the past 20 years, we would expect to see debris from these types of implants to decrease.
Where Does This Leave Materials and Medical Device Developers?
For stakeholders in materials science, medical devices, and occupational health, these findings carry several implications:
Material selection and wear performance: Understanding how wear particles distribute beyond the implantation site may influence material choices and design strategies for implants and other long-term devices.
Exposure assessment in high-risk settings: Industries involving fine plastic particulates, thermal processing, or high-energy machining may benefit from targeted tissue or biological fluid screening for micro- and nanoplastics.
Regulatory and toxicological frameworks: As tools like Py-GC/MS make it possible to characterize internal plastic burdens more accurately, regulatory expectations and risk assessment paradigms are likely to evolve.
Exposure appears to be inevitable: Although the Nihart study is for a relatively small population, the data suggests that no one is immune to this risk, and consideration of the reduction of single use applications and more efficient recycling is clearly going to be of importance.
Cambridge Polymer Group supports this emerging area with Py-GC/MS-based micro- and nanoplastic assays for biological and environmental matrices, as well as complementary materials characterization services. If you are evaluating plastic exposure in a high-risk population, characterizing wear debris from a new device, or designing a toxicology study that requires polymer-specific quantitation, our team can help you develop an appropriate analytical strategy.
To discuss a specific application or study design, please contact us to speak with one of our scientists.
[1] Nihart, A. J., et al. (2025). “Bioaccumulation of microplastics in decedent human brains.” Nature Medicine., Published 2/3/25, DOI 10.1038/s41591-024-03453-1
Posted by rpm rpm on | Comments Off on Dummy Post: To Check Mail, That Is the Question #2
Our chromatography team is regularly asked to identify compounds in materials. Some projects only require the identification of compounds, while others require the accurate determination of concentration of the identified compounds. The latter, termed quantitative chromatography, requires the preparation of calibration standards suitable for the compound in question. At a recent dinner, one of our scientists recognized, in an acute way, the benefit of concentration assessment over the more qualitative assessment of presence for the compound capsaicin.
Posted by Dr. Stephen Spiegelberg on | Comments Off on Caliente Chromatography: Quantitative Analysis of Capsaicin
Chromatography: To Quantify or Not to Quantify, That Is the Question
Our chromatography team is regularly asked to identify compounds in materials. Some projects only require the identification of compounds, while others require the accurate determination of concentration of the identified compounds. The latter, termed quantitative chromatography, requires the preparation of calibration standards suitable for the compound in question. At a recent dinner, one of our scientists recognized, in an acute way, the benefit of concentration assessment over the more qualitative assessment of presence for the compound capsaicin.
Why Tails Matter
Molecular Tweaks That Determine Whether You’re Enjoying Salsa or Enduring Spider Toxins
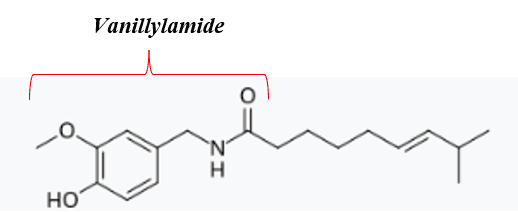
Capsaicin, or 8-methyl-N-vanillyl-6-nonenamide, is the compound found in chili peppers that provides the burning sensation in tissues with which it comes into contact. Capsaicin is the predominant compound found in the general category of capsaicinoids, shown in Figure 1. These compounds all have the same vanillylamide structure at one end of the molecule (left in the figure) but have differing aliphatic tails. Interestingly, the vanillotoxin of the venom of some tarantulas activate the same pain pathways as capsaicin, although arguably through a much less enjoyable mechanism.
Figure 1: Capsaicin compound. Common Vanillylamide group marked on the left with the unmarked tail on the right varying between compounds.
The Scoville Heat Unit
Consumers of food prepared with chili peppers instinctively know that there are levels of heat in the food that is dependent on the concentration of capsaicin. Around 1912, William Scoville, a pharmacist, developed a subjective scale to provide a ranking of the hotness of chili peppers. In his original test, a known weight of dried pepper was extracted in ethanol and diluted to specific concentrations of solutions of the extract in sugar water.
These solutions were tasted by a panel of tasters until a majority could no longer detect the heat in the solution. The heat unit was based on the amount of dilution necessary to lose the detectable heat, so a pepper requiring a dilution of 1 million times before no heat was detected would have a Scoville Heat Unit (SHU) of 1 million.
The hottest pepper tested to date is Pepper X with a SHU of 2,693,000, created in 2023 by crossbreeding the Carolina Reaper with another pepper. For comparison, a Jalapeno pepper has an SHU between 2,500-10,000.
Scoville aimed to assess capsaicin content for use in muscle salves and related pharmaceutical products. His scale became important for both culinary and pharmaceutical applications, especially in ensuring product consistency, safety, and efficacy.
Replacing Tongues with Chromatography Columns
An obvious weakness of this subjective technique is variability between tasters and sensory fatigue. To remove this subjectivity, the assessment of SHU is now performed with high performance liquid chromatography, which can quantitatively measure the concentration of capsaicinoids directly. Dried pepper samples are extracted in acetonitrile and the peak area associated with the capsaicin compound is compared to calibration curves prepared from standards. The results are free from subjective bias, and the test subjects are much happier. This approach is very similar to the trace compound analysis performed by Cambridge Polymer Group.
Capsaicin Quantification Applications
Food Industry: Manufacturers of spicy foods, sauces, and extracts need precise capsaicin quantification for flavor standardization, product labeling, legal compliance, and quality assurance.
Pharmaceutical/Medical Device: Companies developing capsaicin-containing topical creams, patches, or other formulations require accurate quantification during R&D, regulatory submission, and quality control for clinical use.
Packaging: In modern packaging science, capsaicin is directly incorporated into biomaterials such as antimicrobial films, coatings, and encapsulation systems to enhance freshness, control microbial growth, and improve barrier properties for food and medical products.[1]
Capsaicin quantification is crucial not just as a safety and contamination measure, but as a technical quality parameter for new biomaterial products and advanced controlled-release systems. These advanced delivery systems still depend on robust quantitative chromatography to verify capsaicin loading, release profiles, and shelf-life performance in real formulations.[2]
Contact Cambridge Polymer Group today for expert chromatography, quantification, and material characterization services. Speak to one of our scientists to help define your product or research needs.
[1] Qincong Luo, Jinyu Ouyang, Luqi Zhan, Guohuan Liang, Xiaojuan Wang, Development and characterization of capsaicin-enriched Dialdehyde starch-PVA films for antimicrobial food packaging. International Journal of Biological Macromolecules.2025;330(Pt 1):147918. doi: 10.1016/j.ijbiomac.2025.147918.
[2] Qiu X, Xie J, Mei J. Recent Advances in the Applications and Studies of Polysaccharide-, Protein-, and Lipid-Based Delivery Systems in Enhancing the Bioavailability of Capsaicin-A Review. Polymers (Basel). 2025 Apr 27;17(9):1196. doi: 10.3390/polym17091196. PMID: 40362978; PMCID: PMC12073809.
Posted by Dr. Stephen Spiegelberg on | Comments Off on How Long Can a Polymer Last…And What Is Q10?
The longevity of polymers in real-world use is a critical importance across industries, from medical devices and packaging to consumer products and infrastructure. While many polymers are engineered to withstand sunlight, heat, moisture, and chemical exposure, nearly everyone has seen a once-flexible product turn brittle, discolored, or cracked over time. Both raw materials (such as pellets or powders) and finished products can slowly degrade in storage or use, leading to cosmetic changes or even complete functional failure.
Because real-time testing of decades-long product lifespans is rarely practical, manufacturers rely on accelerated aging studies to estimate a polymer’s shelf life and in-use performance. These tests subject materials to elevated stress conditions that replicate the effects of long-term aging within a much shorter timeframe, providing scientifically grounded predictions of stability.
Accelerated Aging and the Arrhenius Equation
A common approach to accelerated aging is through heat, exploiting the known exponential relationship between temperature and reaction rates described in the Arrhenius equation, as shown in Equation 1, where k is the reaction rate, E is the activation energy for the reaction, T is the temperature of the storage environment, and R is the universal gas constant (8.314 JK-1mol-1). A is a prefactor term that is usually determined empirically.
Equation 1
In an accelerated aging study, the researcher establishes a property that will be affected by aging and then sets a threshold limit on that property where the material or product no longer meets its specifications. Experiments can then be conducted at various temperatures over time to determine the time to failure at these temperatures. Equation 1 can be re-written in terms of time to failuret1at the accelerated temperature T1relative to the real time conditions (time to failure t0 at real-time temperature T0) as:
Equation 2
By determining reaction rate at two different temperatures, the A prefactor is not needed and the only unknown in these equations is the activation energy. This parameter is a function of the material and how it was processed and describes the minimum amount of energy required to initiate a chemical reaction. There are a few ways of experimentally determine the activation energy (or in some cases tabulated values can be used).
Determining Activation Energy
ASTM D3045 Approach
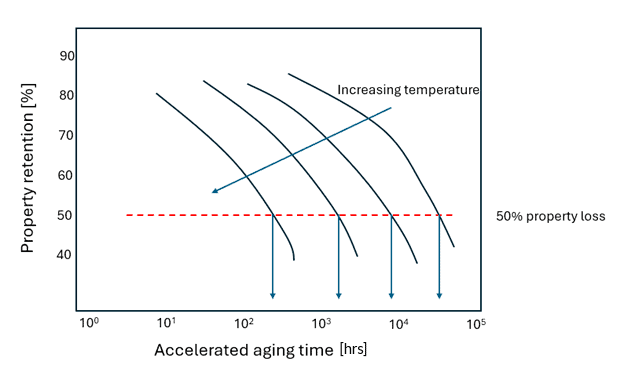
In one method, samples are aged at four or more temperatures for varying amounts of time, and the property of interest, such as tensile strength, is measured for each of these specimens over time (see Figure 1 and Figure 2).
Figure 1: Property loss as a function of temperature following ASTM D3045 at four different temperatures. The related time to failure values are noted with the downward facing arrows, which are used in Figure 2.
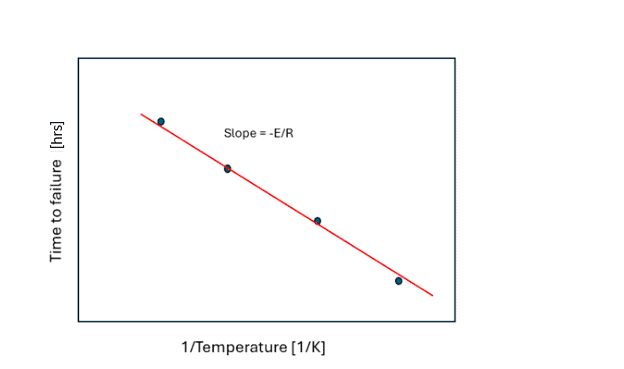
Figure 2: Determination of activation energy, E, from Figure 1.
A threshold property value is used to plot a time versus inverse temperature plot (Figure 2), the slope of which yields activation energy. This method is a reliable way of determining the activation energy, which can then be used for assessing property loss at different storage temperatures. The downside of this approach is that many experiments (and time) are required to build a dataset.
ASTM E1641 Approach
A quicker approach is to determine the activation energy through ASTM E1641, where a set of thermogravimetric analyses (using a Thermogravimetric Analyzer, or TGA) at different heating rates are used to monitor thermally-driven mass loss. In a simple system, the thermally-driven mass loss is an indication of the resistance of the material to thermal decomposition, and the heating rate provides the kinetic driver for the testing. By testing four samples at different heating rates and selecting a target mass loss (say 5%), a plot of log (heating rate) vs. (1/T), similar to Figure 2, will be generated, with the slope proportional to the activation energy. However, it should be noted that in our experience, the calculated activation energy of these two techniques is rarely identical.
Dynamic Rheology / Master-Curve Approach
There is a third common technique for polymers. The dynamic rheological properties of a polymer also generally obey the Arrhenius function and therefore building a so-called master-curve from a set of rheological experiments at different experiments. The shift factor used to create this master-curve is related to the activation energy of the polymer, providing another way to estimate this important parameter.
Simplifying with the Q10 Factor
Discussing aging rates in terms of activation energies can be unwieldly. As a result, researchers performing accelerated aging studies often refer to the Q10 value, which is the pre-factor that considers how much the reaction rate Rn with an increase of 10 ºC. The Q10 factor is simply written as:
Equation 3
where R1 and R2 are the reaction rates at the respective temperatures. The Q10 can be determined from the activation energy through manipulation of Equation 2 and Equation 3. Knowing a Q10 value for your material or product, you can quickly see real time (RT) shelf-life equivalent values based on accelerated aging temperatures (A) by rearranging Equation 3 to get:
This process is captured in ASTM F1980. As an example, for a Q10 of 2.0, accelerated aging for 30 days at 50 ºC relative to real time storage conditions at 23 ºC would give a real time equivalence of 195 days. Often this value of Q10 is used as a default value, but selection of the accelerated aging temperatures, and calculation of an accurate Q10, requires detailed knowledge of the materials used.
Learn More in Our Accelerated Aging Webinar
To explore these concepts in more depth, including practical limitations and real-world examples, attend our webinar:
Age ISN’T Just A Number: Accelerated Aging Methods for Material and Product Characterization Date: Wed, Jan 21, 2026 Time: 2:00 – 3:00 PM EST
In this presentation, we will discuss accelerated aging as a ubiquitous tool used to ensure shelf and in-use stability over long time periods. This powerful collection of techniques allows prediction of potential aging issues that would be time consuming and costly to identify in real time. However, accelerated aging has limitations, and data generated in an accelerated study must not be used blindly.
CPG Speakers
Gavin Braithwaite, Chief Executive Officer
Jaimee Robertson, Director of Consulting Services
Kalpana Viswanathan, Polymer Chemist
The webinar will:
Review aging mechanisms in polymers and the scientific assumptions that underpin accelerated aging.
Discuss analytical techniques that can be used to screen material aging or infer acceleration factors, including their weaknesses.
Present a case study comparing accelerated aging techniques to demonstrate the importance of careful experimental design when relying on accelerated methods to predict material performance.
Attendees will gain an understanding of the processes underpinning accelerated aging and the key considerations for applying these data to material and product decisions.
Selecting effective appropriate accelerated aging conditions requires a solid understanding of your product’s material composition, processing history, and thermal stability.
Cambridge Polymer Group scientists have decades of experience designing and executing Arrhenius-based and Q10-based shelf-life studies for polymers, medical devices, and specialized materials.
Our lab can help you:
Determine activation energy (via ASTM D3045, E1641, or Dynamic Rheology)
Identify realistic accelerated aging conditions
Validate shelf-life claims and packaging performance
Contact us to design an accelerated aging study tailored to your material, product, and regulatory needs.
Posted by Catherine Cerasuolo on | Comments Off on I Have a Little Robot
I have a little robot, For minimally invasive care, With polymer housings, cable sheaths, And coatings tuned for wear.
It moves by catheter guidance, With robotic hands outside, The console shapes its pathway While surgeons steer inside.
A handheld little robot Helps line the cuts just right, It tracks the plan from CT scans For guided implants, watertight.
Oh robot, robot, robot, With torque in every arm, Your motion’s finely planned and checked To help reduce the risk of harm.
Oh robot, robot, robot, Your gears and bearings glide, With clear control and smooth response So surgeons trust you at their side.
CPG checked your polymer housings, Your coatings, strain, and wear, Ran aging, creep, and fatigue tests To make sure you’re stable in there.
They tested contact surfaces early, For debris and surface wear, Characterized particulates and flakes To keep loose matter rare.
They mapped out stress and fatigue life, From guidewires to soft grips, And added sterilization cycles To see what really slips.
Then chemists chased extractables, With leachables in view, To give tox teams the biocomp data For risk on what those chemicals might do.
They wrapped it all in reports and tables, Consistent with what regs expect, To support submissions for surgical bots With data reviewers will respect.
Oh robot, robot, robot, Your data tell your tale, CPG’s comprehensive analysis Shows you’re built to work, not fail.
Posted by Dr. Gavin Braithwaite on | Comments Off on 2026: The Year Ahead from Cambridge Polymer Group
In 2026, robotics and AI will continue to receive substantial attention across all industries, including medical devices. The expanding adoption of robotics and AI will coincide with other significant pressures that will impact medical devices. These include the recent 10993-1 update, and how the FDA views this update, as well as ongoing tariff and onshoring pressures. Collectively these forces will reshape how products are designed, validated, and manufactured in the coming year. As a result, in medical devices in particular, a deep understanding of the underlying material science, coupled with robust testing, will become more critical to reduce risk, increase performance, and control time-to-market.
At Cambridge Polymer Group, we have spent decades helping companies navigate the intersection of material science and product innovation in medical devices and beyond. As we look toward 2026, regulatory evolution, robotics, AI, and supply chain dynamics are converging in ways that place materials at the center of product reliability, regulatory success, and commercial viability.
Regulatory Pressures: Complexity in Biocompatibility
The recently revised ISO 10993-1 standard has introduced new complexity and uncertainty into biocompatibility evaluations, while uncertainty surrounding the FDA’s pending recognition leaves many teams in limbo. From our vantage point, this environment emphasizes the need for thorough material characterization and a fundamental understanding of the materials and their properties. Sterilization compatibility, resistance to cleaning agents and disinfectants, additive packages, extractables/leachables, and long-term aging behavior can no longer be addressed as an afterthought. They are central to development and approval timelines, costs, and patient safety.
Robotics and Reliability: Materials at the Core
In robotics, particularly in healthcare, we are seeing a growing awareness that reliability is not just about software or control systems — it is about the materials that bear load, resist fatigue, and survive sterilization and repeated cleaning. Failures tied to creep, environmental stress cracking, and chemical degradation are increasingly traceable to early-stage material mis-selection. Not giving these choices sufficient thought early on can lead to substantial loss of time. More generally, outside of healthcare, we have seen concerns around unexpected wear on wheels, tracks and guides, as well as gear material selection issues. Our work in polymer testing and failure analysis continues to show that the right material, selected with the full performance requirements in mind and validated under realistic conditions, is often the difference between field success and costly redesign.
AI and Materials: Promise and Caution
Recent press on the potential of AI suggests that it may have application throughout the material selection and characterization space. AI has the potential to transform how materials are modeled, selected, and even predicted to fail. In fact, as an example, we have collaborated with clients who use machine learning to discern patterns in complex data sets, simulate polymer behavior, optimize formulations, and accelerate testing. However, the models are only as good as the data used to train them.
The potential constraint on adoption of AI in this space (or the hidden catch in using its output) is that it is rare for published values to include all relevant parameters for material selection. For example, few elastomer data sheets will include information on sterilization compatibility or cleaning resistance, and data on the impact of additives on specific resins is scarce. We therefore caution that AI generated results must be considered from the perspective of the final use-case and must be grounded in physical data. Without robust experimental validation, predictive models risk drifting from reality with heavy consequences — especially in regulated environments like medical devices. CPG is well placed to provide both the fundamental material science and the critical test-based validations of the proposed materials.
Supply Chain Resilience: A Material Challenge Intensified
Finally, trade dynamics, tariffs, and regionalization are adding another layer of complexity to sourcing specialty polymers, medical-grade resins, and advanced composites, which remain heavily globalized and prone to high costs and disruption. We are increasingly seeing suppliers seek second-source vendors or find on-shore (or near-shore) alternatives to support and de-risk existing supply chains. We have also seen firsthand how material substitution under pressure can introduce unexpected risks. That is why we advocate for proactive risk assessment, testing, supplier qualification, and geographic diversification — not just as a procurement strategy, but as a reliability imperative.
Our View
2026 looks to be a year impacted by the convergence of several forces, each of which would be significant on its own and could disrupt companies’ operations if not addressed upfront. For this reason, we feel that 2026 will reward organizations that treat material science as a strategic discipline. Whether it is navigating regulatory ambiguity, designing reliable robotic systems, validating AI-driven insights, or building resilient supply chains, materials are at the center.
How Material Science Connects 2026’s Industry Challenges
Domain
2026 Challenge
Material Science Role
Medical Devices
Regulatory ambiguity around ISO 10993 and sterilization
Validating biocompatibility, sterilization resilience, and long-term aging of polymers
Robotics
Reliability and field failure risks
Selecting materials that resist creep, fatigue, and chemical degradation
AI
Trust, validation, and model drift
Providing physical data to train and validate AI simulations of material performance
Trade & Tariffs
Supply chain fragility and cost volatility
Diversifying sources of specialty polymers and qualifying substitutes under real conditions
At Cambridge Polymer Group, we believe that thoughtful material selection, rigorous testing, and transparent cross-functional communication between engineering, quality, and regulatory teams derisks the development and production pipeline and provides the foundation for innovation that lasts.
Posted by Catherine Cerasuolo on | Comments Off on From Forest Floor to Food Wrap: Turkey Tail Mycelium at the Frontiers of Polymer Science
By early December, most Thanksgiving turkeys have migrated to stock pots and storage containers, but in forests across New England and beyond, turkey tail (Trametes versicolor) spreads over fallen logs in familiar bands of brown and cream. It draws attention less for its resemblance to the holiday bird than for its polymer-rich tissues that enable distinct biological and materials behaviors.
This deceptively simple shelf fungus is packed with complex polysaccharides and mycelial networks that inspire new approaches to immune‑modulating ingredients and bio‑based coatings, right at the intersection of biology and materials science that Cambridge Polymer Group’s clients navigate every day.
Polymers In A Mushroom: PSK, PSP And β‑Glucans
Turkey tail cell walls are rich in high molecular weight polysaccharide-peptides such as PSK (polysaccharide‑K) and PSP (polysaccharopeptide), which combine branched glucan backbones with peptide components. These macromolecules appear to work as a biological response modifier interacting with pattern recognition receptors (for example, Toll‑like receptors) and can modulate cytokine production, natural killer cell activity, and other immune pathways in preclinical and clinical studies.[1][3]
In Japan, a standardized PSK extract from T. versicolor has been used as an adjunct to conventional chemotherapy, and clinical data suggest effects on survival and quality‑of‑life endpoints in several solid tumors. [2] PSP and related fractions are under investigation for similar immunotherapeutic roles and for their ability to influence immune checkpoints and tumor microenvironments.[3]
Gut Microbiome And Prebiotic Effects
Like other fungal polysaccharides, turkey tail fractions behave as fermentable fibers for the gut microbiota, supporting short‑chain fatty acid production and enrichment of beneficial genera such as Lactobacillus and Bifidobacterium in experimental models. Reviews of fungal polysaccharides highlight their potential to modulate gut barrier integrity, systemic inflammation, and metabolic parameters via microbiome shifts, positioning turkey tail as a candidate prebiotic ingredient.[4]
For medical device and drug‑delivery developers, these data illustrate how specific polymer architectures (branching, peptide content, charge) translate into measurable biological responses in mucosal environments. Understanding these structure–function relationships is directly relevant when designing synthetic or semi‑synthetic hydrogels, coatings, and excipients intended to engage the same receptors and tissues.
Mycelium Coatings As Plastic Wrap Alternative
Recent work from University of Maine researchers has shown that turkey tail mycelium, combined with cellulose nanofibrils from wood pulp, can form thin, continuous coatings on paper, textiles, and wood. After several days of controlled growth and a heat‑treatment step, the resulting layer is food‑safe, biodegradable, and resistant to penetration by water, oils, and organic solvents such as n‑heptane and toluene.[5]
This “grown” coating behaves like a bio‑based barrier film, suggesting pathways to replace petroleum‑derived plastic wraps and cup linings. For packaging and materials engineers, it represents a living polymer processing route in which mycelial hyphae and fibrillated cellulose self‑assemble into a functional composite at low temperature and with renewable feedstocks.
Relevance For Medical And Industrial Polymers
From a polymer science perspective, turkey tail offers three complementary case studies.
Immunoactive polysaccharide–peptides illustrate how subtle changes in glycan composition and peptide content shift receptor binding and downstream signaling, informing the design of bioactive coatings, adjuvants, and drug carriers.
Prebiotic effects on the microbiome demonstrate that “inert” excipients can have system‑level consequences, a key consideration for oral devices, controlled‑release matrices, and combination products.
Mycelium–cellulose coatings show how fungal growth can be harnessed as a fabrication step for barrier layers and biocomposites, pointing to future opportunities in sustainable packaging, tissue‑compatible substrates, and low‑impact foams.
As companies look to align product development with circular‑economy and ESG goals, bio‑derived polymers like those from Trametes versicolor highlight how materials design, biology, and regulatory science intersect. Cambridge Polymer Group can support clients in this space through characterization of bio‑based coatings and composites, structure–property testing of novel polysaccharide systems, and guidance on test strategies for biocompatibility and degradation under relevant standards.
[1] Standish LJ, Wenner CA, Sweet ES, et al. Trametes versicolor mushroom immune therapy in breast cancer. J Soc Integr Oncol. 2008;6(3):122–128. Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC2845472/
[2] PDQ Integrative, Alternative, and Complementary Therapies Editorial Board. Medicinal Mushrooms (PDQ®): Patient Version. National Cancer Institute; updated July 11, 2024. Available at: “How Two Document Examiners Solved the Case of the Salamander Letter.” https://www.ncbi.nlm.nih.gov/books/NBK424937/
[4] Barcan AS, Barcan RA, Vamanu E. Therapeutic potential of fungal polysaccharides in gut microbiota regulation: implications for diabetes, neurodegeneration, and oncology. J Fungi. 2024;10(6):394. doi:10.3390/jof10060394. Available at:https://pmc.ncbi.nlm.nih.gov/articles/PMC11204944/
[5] Zier S, White LR, Johnstone D, et al. Growing sustainable barrier coatings from edible fungal mycelia. Langmuir. 2025;41(39):26751–26759. doi:10.1021/acs.langmuir.5c03185. Available at: https://doi.org/10.1021/acs.langmuir.5c03185
Posted by Dr. Stephen Spiegelberg on | Comments Off on The Chemistry Behind the Perfect Roast: Understanding the Maillard Reaction
Every time you roast a turkey or bake bread, a fascinating chemical reaction gives your food its rich brown color, enticing aroma, and complex flavors. That reaction is called the Maillard reaction (pronounced my-ard), a cornerstone of both food chemistry and polymer science.
What Is the Maillard Reaction?
The Maillard reaction occurs when amino acids (from proteins) react with reducing sugars at temperatures above about 140 °C. It is not a single reaction, but a chain of them that unfolds in three general stages: initial, intermediate, and final.
Initial reaction: Carbonyl groups from sugars react with amino groups from amino acids to form Amadori products, also known as glycated proteins.
Intermediate reaction: These products break down into smaller molecules such as reduced sugars, dicarbonyls, and additional amino compounds.
Among the many products of the Maillard reaction are melanoidins, a group of high-molecular-weight nitrogen‑containing polymers that give foods their brown hues. Their structures vary depending on the starting sugars and amino acids but generally include heterocyclic rings such as pyrroles, furans, and pyridines linked to a carbohydrate backbone.
These same compounds are responsible for the characteristic color of roasted coffee, seared steaks, and baked bread. Melanoidins and other Maillard products also create the familiar flavor molecules that chefs prize:
Pyrazines: roasted or toasted notes
Thiophenes: rich, meaty flavor
Furanones and furans: sweet, caramelized aroma
Oxazoles and pyrroles: nutty or sweet nuances
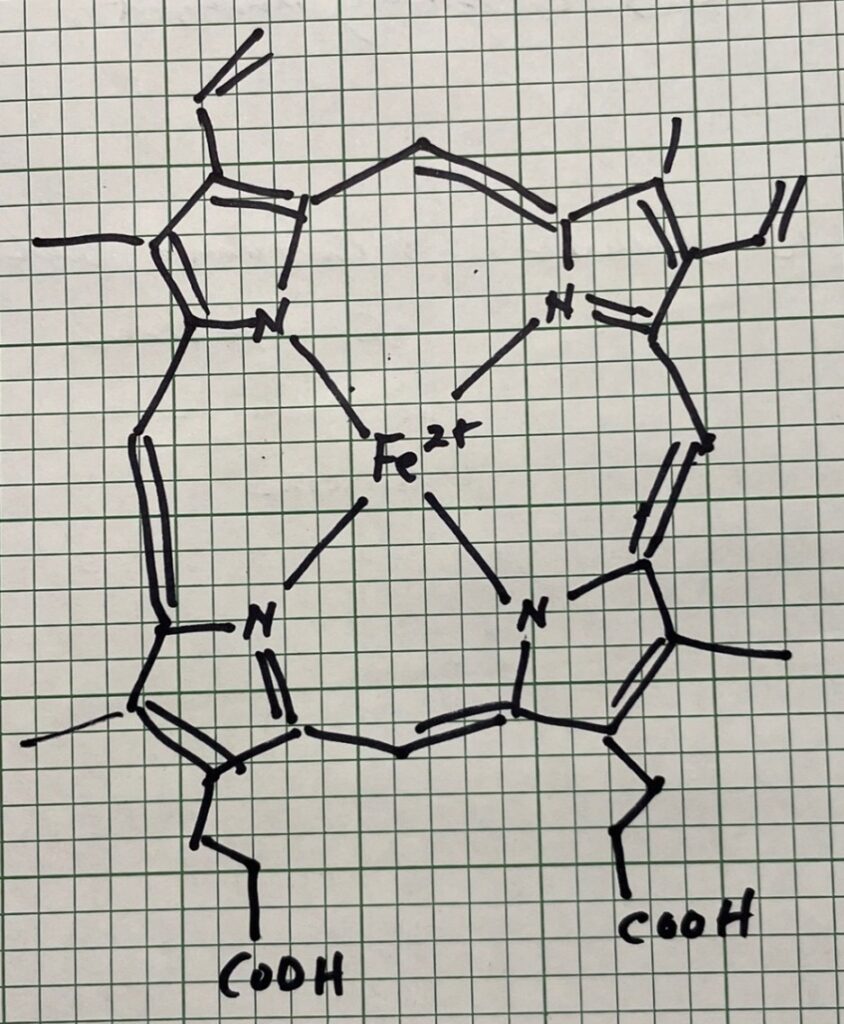
Beyond the Maillard Reaction: Myoglobin and Color
Not all browning in cooked meat comes from the Maillard reaction. Another source of the brown color is myoglobin, composed of 150 amino acids found in the muscle tissue of vertebrate animals (see Figure 2) and is used to store oxygen in muscles. Myoglobin has four pyrrole nitrogens that surround a ferrous ion center, as shown below. As meat heats, the heat denatures the protein and the myoglobin. This transition occurs much lower than the Maillard reaction (of the order of 60 °C) and is the driving force for the bulk color change that allows determination of “doneness” in red meat since the form of the converted myoglobin governs the final color of the molecule.
Figure 2: Myoglobin
How the Maillard Reaction Differs from Caramelization
The Maillard reaction should not be confused with caramelization, which involves the direct pyrolysis (thermal breakdown) of sugars without amino acids. Both processes create brown color and complex flavors, but they arise through distinct chemical pathways.
Let’s Talk Turkey: Maximizing the Maillard Reaction
When it comes to roasting a turkey, harnessing the chemical reactions involved in cooking makes all the difference in flavor and appearance. Here are a few science-backed tips:
Dry the surface. Removing excess moisture by blotting helps the turkey brown more quickly.
Use moderate alkalinity. Raising the surface pH with a small amount of baking soda encourages the Maillard reaction.
Maintain high heat. A roasting temperature above 140 °C ensures the reaction proceeds effectively.
Control heating rate and conditions. Although not relevant for surface browning, the rate of heating, and the oxygen environment (oven versus barbeque) can impact the color of the meat in the way that the myoglobin is degraded.
So do you need to be a chemist to cook a turkey? Thankfully, the answer is no, as all these reactions occur naturally just by cooking the turkey at the appropriate temperature and for the appropriate duration of time. However, when the author of this blog post was a graduate student in MIT’s Chemical Engineering department, the department secretaries received several phone calls every November from amateur chefs asking questions about how to cook their turkeys. The standard response from the secretaries? “Let us connect you to the Chemistry department.”
Posted by Catherine Cerasuolo on | Comments Off on ASTM F04.15.17 Workshop Highlights Advancing Standardization in Medical Device Cleaning
The ASTM Committee F04.15.17 on Medical Device Cleaning recently held a workshop focused on the analysis of cleaning agents used for both new and reusable medical devices. The goal was to identify key topic areas requiring standardization to help ensure the development of safe, effective, and well-characterized products across the medical device industry.
The morning sessions opened with an overview of current standards for clinically used devices, such as endoscopes and surgical instruments, presented by Ralph Basile (Healthmark). Subsequent talks explored strategies for identifying manufacturing residues, addressing worst-case cleaning challenges, and defining product grouping considerations, with contributions from Ben Grosjean (Zimmer), Ramanthan Dhakshinamoorthy (Procept BioRobotics), and Sarah Frank (Johnson & Johnson).
Jeff Phillips (Alconox) and David Ruiz (Agilitti Health) led discussions on detergent composition and its impact on cleaning performance. Several presenters then examined analytical methods for evaluating cleaning agent residues, including:
Analysis of volatile compounds by Brian Bosso (Steris)
Spectroscopic techniques by Mayuri Kasareni (Intuitive Surgical)
Chromatography methods by Stephen Spiegelberg, Mimoza Xheka, and Becky Bader (Cambridge Polymer Group)
Alex Freeman (Intuitive Surgical) and John Howell (Novonesis) discussed how cleaning agents interact with biological tissues, while Rob States (Cormica) presented case studies illustrating failure analyses linked to improper cleaning protocol implementation.
The workshop concluded with a panel discussion led by conference chairs Alpa Patel, Kaumudi Kulkarni, and Barbara Kanegsberg, summarizing key takeaways and identifying areas for future standardization work.
Key Insights and Next Steps
Detergent efficacy testing: A standardized approach is needed to enable meaningful comparison of cleaning agent performance.
Unknown composition risks: Common test methods and risk assessment frameworks should be developed to address cleaning agents with partially disclosed or proprietary formulations.
Healthcare facility engagement: Greater collaboration with healthcare providers is essential to ensure that clinical cleaning processes meet appropriate standards. Currently, most healthcare facilities are not involved in ASTM or ISO initiatives focused on cleaning and are not subject to FDA regulation in this area. The committee plans to explore ways to involve this community more directly in future standardization efforts.
Posted by Catherine Cerasuolo on | Comments Off on Why Getting Material Selection Right Matters in Medical Device Design Live Event
Selecting the right material from day one can make or break a modern medical device.
Join Cambridge Polymer Group for “Getting Material Selection Right the First Time” with industry leader Dr. Gavin Braithwaite on November 12, 2025, at 2:00pm EST.
Medical device development today is a balancing act. Teams must juggle evolving regulations, rapid market shifts, and manufacturing constraints, all before the device ever leaves.
Relying solely on conventional specification sheets or historical precedent carries serious risks, especially as design iterations speed up and requirements become more complex. Dr. Braithwaite will use orthopedic implants as a historical lens to show how early material decisions impact long-term safety and performance.
Pressures Shaping Medical Device Design
Material selection isn’t just about picking what’s familiar. Teams must consider:
• New regulatory hurdles, such as PFAS restrictions in the EU.
• Market pressures, such as on-shoring or additive changes.
• Changing sterilization trends, including demand for alternatives to ethylene oxide.
• The impact of miniaturization on material integrity and performance.
• The novel challenges posed by degradable polymers, hydrogels, and bio-compatible formulations.
Avoiding Costly Mistakes
Insights from Cambridge Polymer Group’s consulting work highlight recurring pitfalls:
• Failing to involve material science experts early in development can lead to delays, recalls, and design reboots.
• Overlooking the value of early material vetting leaves smaller companies exposed to unforeseen costs.
• Horror stories abound of late-stage changes forcing teams to restart validation or compromise time-to-market.
Industry Voices: Best Practices for Selection
Drawing on industry best practices:
• Material selection must begin at the earliest stages, not after a prototype is built.
• Design teams must collaborate closely with regulatory, manufacturing, and materials science specialists.
• Consider a material’s regulatory status, biocompatibility, mechanical properties, and manufacturability together, not in isolation.
• Maintain flexibility; if forced to pivot late, engage cross-functional teams and expert partners to minimize risk.
Cambridge Polymer Group works with OEMs of all sizes to:
• Anticipate and navigate regulatory and market changes.
• Advise on specialist materials for degradable, hydrogel, and combination devices.
• Streamline material vetting, ensuring projects stay on timeline and budget.
Register for the Live Event
Tap into Cambridge Polymer Group’s expertise and join Dr. Braithwaite for actionable guidance on November 12. Get ahead of the curve and learn how leaders in the industry approach “Getting Material Selection Right the First Time.”
Reserve your seat and be prepared to ask the questions that will get your device to market, fast, safe, and compliant.