Posted by Catherine Cerasuolo on | Comments Off on Bridging the Gap in Material Science Expertise: Explore the New Campoly.com
We are excited to unveil the new campoly.com, the redesigned digital home for Cambridge Polymer Group, Inc. (“CPG”)! This launch marks a major milestone in our commitment to providing advanced contract testing, research, and development services in material science for clients across all industries, with particular emphasis on healthcare.
Why We Redesigned
Over the past 30 years, CPG has grown into a trusted partner for clients spanning medical devices, consumer products, industrial manufacturing, and beyond. Our expanding material science expertise demanded a website that reflects both the depth of our team’s experience and the breadth of our capabilities. The new campoly.com is designed to offer a seamless experience for prospective and returning clients, researchers, and industry partners.
What’s New on campoly.com
Improved Navigation: The new site’s intuitive menu structure makes it easy to find information about our contract research, material development, and testing services.
Expanded Resources:Explore our content across all our disciplines and navigate through our updated blog and publications to stay informed on the latest materials science trends and innovations. No paywalls here!
Team & Expertise: Get to know CPG’s world-class team of PhD scientists and engineers and discover how our subject matter experts guide complex polymer-based projects from concept to commercialization.
Client-Centered Support: Quickly access details on submitting samples, requesting quotes, or collaborating on custom projects, with direct links to our contact page and streamlined forms.
Enhanced Client Collaboration
Our new website embodies Cambridge Polymer Group’s client-first approach by making our suite of services more accessible than ever. As we continue to innovate in materials development, testing methodologies, and scientific consulting, the revamped campoly.com serves as an essential touchpoint for project inquiries and knowledge sharing.
Have questions, or want to discuss your next project? Reach out via our contact page—we’re ready to help accelerate your success through polymer science.
Posted by Dr. Stephen Spiegelberg on | Comments Off on From Residues to Risk: Why Medical Device Cleaning Validation Matters in Biocompatibility Assessments
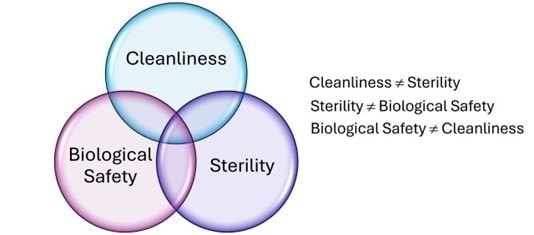
Ensuring medical device safety requires a coordinated approach across three critical domains: biocompatibility, cleanliness, and sterility. While each area has its own regulatory and testing requirements, they are deeply interconnected. Overlooking the relationship between these elements can increase patient risk and delay product approvals. This upcoming medical device webinar explores how cleaning validation influences biocompatibility outcomes and overall product safety.
Cleaning validation is a foundational step in establishing device safety. Surface contaminants left from manufacturing or cleaning processes can directly impact biological evaluation results and compromise sterility assurance. Similarly, certain sterilization methods can alter material chemistry, generating new extractables or leachables that affect the device’s biocompatibility profile.
When designing and validating a medical device, both manufacturers and regulators should consider:
Cleanliness addresses surface residues, which can directly influence biological evaluation results. It’s also essential to achieving reliable sterilization assurance.
A sterile medical device may not necessarily be biocompatible; sterilization methods can alter chemical properties relevant to biological safety.
Devices can meet cleaning and sterility standards but still fail biocompatibility due to material selection or design factors.
Even if a device is determined to be biocompatible post-manufacture, poor cleaning or sterility assurance can compromise clinical performance and patient safety.
Join our webinar, From Residues to Risk: The Role of Cleaning Validation in Biocompatibility Assessments, on October 15, 2025, 2 p.m. ET, to learn how cleanliness, sterilization, and biocompatibility testing intersect during medical device validation. Our experts will highlight best practices for integrating cleaning validation into biocompatibility assessment plans, review regulatory expectations, and provide actionable strategies to minimize risk while ensuring compliance with ISO 10993 and related standards.
Posted by Dr. Stephen Spiegelberg on | Comments Off on Hydrogel Water Beads: For Farming Use Only
What Are Water Beads?
“Water beads” are water-absorbent polymer beads that can swell over several hundreds of times their initial dry mass when placed in water, forming hydrogel beads. They are primarily marketed as agricultural products to act as humectants in soil, helping retain and slowly release water into the soil.
Beyond farming, however, water beads have been marketed as colorful sensory toys for play and decoration. This non-agricultural use has raised safety concerns, especially when the beads are accessible to young children who may accidentally ingest them.
What Are the Hazards of Water Beads?
A 2025 case study, published in Pediatrics, documented a 13 month-old who was admitted to the emergency room after a 12 hour stint of vomiting and lethargy[1]. She presented as dehydrated, abdominal distension and tenderness. A radiograph and ultrasound showed a mass in the small intestine, which a laparotomy confirmed to be swollen fragments of water bead material.
The mass was removed, which resulted in partial recovery. The patient continued to show developmental symptoms, and 9 months after the initial mass was removed, a colonoscopy revealed inflammation of the ascending and sigmoid colons and additional gelatinous fragments consistent with water bead material. The patient’s motor skills and speech skills required early childhood intervention. This case underscores the risk of intestinal obstruction and possible neurotoxic effects associated with water bead ingestion.
What Are Water Beads Made Of?
The study author noted that some manufacturers claim the sensory toy water beads are made exclusively of sodium polyacrylate, which is the salt form of polyacrylic acid, a common hydrogel often used as the sorbent in diapers. However, characterization testing performed by Cambridge Polymer Group scientists showed that commercially-available water beads procured by the author and sold as sensory toys, found they were predominantly comprised of polyacrylamide, which is synthesized from acrylamide monomer. Polyacrylamide has a different toxicological risk profile than polyacrylate and hence needed additional scrutiny for its use as a toy.
What Is the Current Regulation on Water Beads?
The lead author of the 2025 study collaborated with the Consumer Product Safety Commission (CPSC) to highlight the risks posed by these products. The CPSC’s own testing of 14 water bead toy products revealed:
Detectable residual acrylamide monomers in most products
Two products exceeding acute oral minimum risk levels
Significant batch-to-batch variability in monomer content
To protect consumers, the CPSC launched public guidance warning parents not to allow children to play with water beads[2]. On August 21, 2025, the Commission approved a new federal safety standard regulating water bead toys. The standard imposes two major restrictions:
Limits on permissible concentrations of polyacrylamide
Rules on maximum allowable bead size when fully swollen to reduce choking and intestinal obstruction risks[3]
What Is Next?
Water beads may have benefits in farming, but as toys, they present serious risks. With new safety standards in place and ongoing research, regulatory science is catching up to protect the most vulnerable consumers.
At Cambridge Polymer Group, we continue to work with companies to characterize the chemical composition of hydrogel materials in consumer products. Our goal is to help ensure these materials are safe, compliant, and effective—whether in agricultural settings or other approved applications.
If your company is working with hydrogels—whether for consumer products, agriculture, industrial, or medical applications—Cambridge Polymer Group can help. From material characterization and performance testing to product development and risk assessment, our scientists provide the insights you need to innovate with confidence. Connect with us to see how we can support your next project.
[1] Haugen A, Friedman E, and Duff I. Intestinal Obstruction and Neurotoxicity Associated With Water Bead Ingestion. Pediatrics. 2025;155(2): e2023065575
When a product unexpectedly fails, performs below expectations, or does not meet safety standards, the cause often lies deep in the materials—or even in trace contaminants—you never knew were there. That’s where material deformulation becomes essential. Rather than building a product from scratch (formulation), deformulation is the investigative process of analyzing an existing material to reveal all its ingredients: base polymers, additives, surface treatments, and even subtle residues from manufacturing.
This deep analysis serves several vital roles: it helps identify the sources of product failure, clarifies why a competing product works better, and offers confidence when switching suppliers or moving manufacturing in-house.
Why Deformulation Matters
Reveals sources of product failures and performance issues, leading to actionable troubleshooting and improvement.
Allows teams to benchmark their products against competitors and discover what drives superior performance elsewhere.
Validates the composition of materials for regulatory compliance and safety assurance.
Enhances quality control by unveiling undisclosed changes or process-introduced impurities.
Supports innovation and cost optimization by uncovering reformulation opportunities or substitute ingredients.
What You’ll Learn
This webinar will introduce the fundamentals of material deformulation—what it is, why it matters, and how it empowers manufacturers to tackle complex material problems. Attendees will discover the main analytical tools of deformulation]. The session will explain how these techniques lead to a comprehensive breakdown of materials and how the resulting insights can help teams reduce risk, troubleshoot failures, design safer products, and improve consistency.
Real-World Case Studies
Dr. Kalpana Viswanathan will share examples of how deformulation made a tangible difference for clients:
Uncovering a Mystery Ingredient: Identifying and characterizing a hidden component in a processing aid, which enabled clearer specifications and better performance in future production.
Benchmarking Against a Competitor: Diagnosing the cause of superior tensile strength in a competitor’s sample, including differences in crystallinity, fillers, and processing—leading to recommendations for improvement.
Solving Safety Testing Failures: Tracing a failed cytotoxicity test to a specific contaminant, troubleshooting its source, and helping the client implement an effective mitigation plan.
Understanding Syringe Cracking: Identifying unreacted epoxy monomer and process variability as root causes of product defects, along with best practices for handling and assembly.
About the Speaker
Dr. Kalpana Viswanathan is a seasoned polymer chemist with over ten years of research and development experience. Her portfolio includes pioneering coatings for implantable medical devices and recent work supporting medical device materials. She holds a U.S. patent in plasma bonding technology and is experienced in hydrogel synthesis, surface modification, biocompatible materials, and comprehensive material characterization. Dr. Viswanathan’s work bridges cutting-edge scientific discovery and practical problem-solving for safer, more effective products.
Why Attend
Whether you’re in medical devices, pharmaceuticals, or consumer products, hidden material risks can undermine your product just when you least expect it. This webinar will demonstrate how material deformulation unveils the real causes of product challenges and equips teams with practical solutions.
Register today to secure your spot and gain practical strategies for uncovering the unseen in your products.
Posted by Joseph White, Ph.D. on | Comments Off on There’s More to Cutting Tissue Than You Think
In medical device development and surgical training, having a suitable test environment is essential for screening device concepts and reliably training surgeons. Traditionally, this process relied on excised animal or human cadaveric tissue—but the use of such tissue is problematic from both ethical and practical perspectives, including issues with stability and variability.
Hydrogel systems now offer a promising alternative: they are ethical, reliable, and mass-producible tissue models, thanks to their wide range of potential material properties. Yet, matching hydrogel properties to natural tissue is no trivial feat. Consider the simple act of cutting tissue with a scalpel: what mechanical properties are needed to achieve the correct cutting “feel”?
The Hidden Complexity of Tissue Cutting
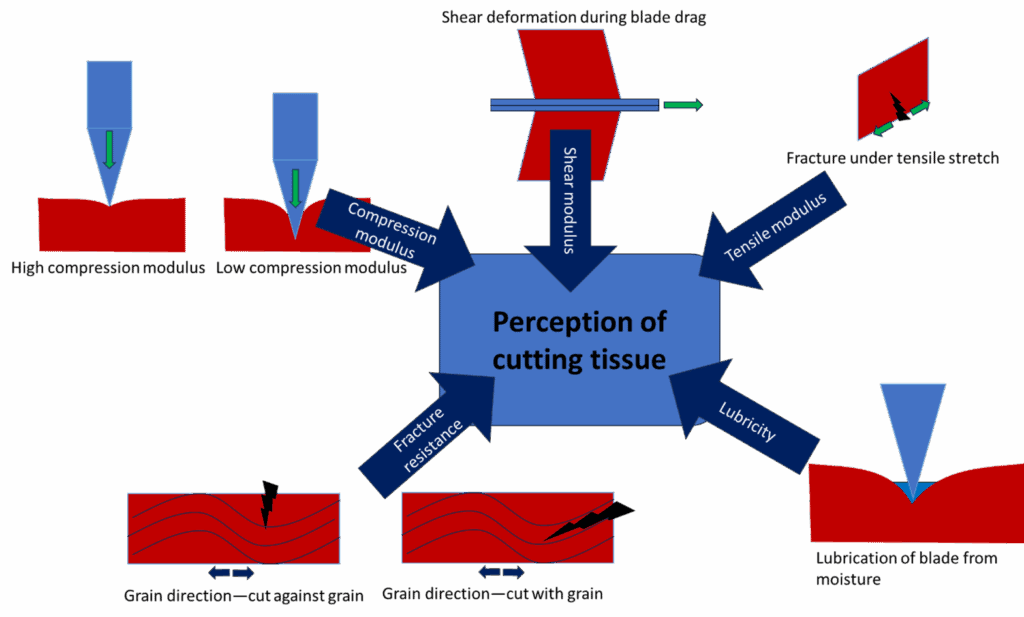
Cutting tissue with a scalpel is a surprisingly complex process, involving a delicate balance of mechanical and rheological behaviors, as well as the natural inhomogeneity of tissue. Because many tissues and organs are non-uniform, accurate mechanical testing is challenging. The most common property reported in the literature is modulus—typically in compression—but this alone cannot capture the complexity of real tissue.
Cutting is fundamental to surgical procedures, whether using a scalpel or more advanced techniques like radiofrequency ablation, laser cutting, or electrocautery. To ensure effective surgical training and device prototyping, it is crucial to replicate the appropriate “feel,” or psychorheology, of cutting natural tissue.
Beyond Modulus: The Role of Fracture Resistance and Lubricity
When a scalpel cuts into tissue, modulus is important, but it is not the only factor influencing the cutting experience. The process begins with the compression of tissue under the blade, followed by the slicing motion and ultimately the separation of tissue. The initial contact is characterized by the compression modulus: soft tissues like lung or brain may be difficult to cut due to excessive compression, while firmer tissues like cartilage or skin provide a more stable feel.
However, compression modulus alone is insufficient to predict or mimic the cutting experience. Another critical factor is the tissue’s resistance to stretching and tearing. Imagine cutting through chicken or steak: as the blade moves, the tissue is stretched (in tension and shear) until it fractures. The cutting feel depends on whether the cut is made with or against the grain.
Muscle tissue, for example, contains a fibrous network. Along the fiber direction, it is harder to stretch; perpendicular to the fibers, stretching is easier. Compression, tensile, and shear moduli all contribute to the overall cutting feel.
Cutting with the grain is generally easier, as the blade can slip between fibers to slice through softer tissue, which has lower fracture or tear resistance. Most natural tissues have a fibrous network that prevents catastrophic tear propagation—this is crucial, as it means small nicks do not become life-threatening tears.
Most natural tissues are moist and/or contain oils. The lubricity—how easily the blade moves—is affected by the presence of fluids like blood or fatty oils. If tissue sticks to the blade, the cutting action can feel dull or draggy. Lubricity is therefore another key component of the cutting experience and can be quantified through tribological measurements of the coefficient of friction in the presence of relevant lubricants.
Overview of “psychorheological” components of simple cutting of tissue.
The Challenge of Mimicking Natural Tissue
As this overview shows, even the simple act of cutting with a scalpel depends on many interrelated factors. The blade’s shape, surface energy, and roughness also play roles, but here we focus on the tissue itself. The complex structure of natural tissues is difficult to replicate with hydrogels, despite recent advances in achieving a wide range of stiffness values.
To truly mimic the feel of cutting natural tissue with a synthetic hydrogel material, it is essential to recognize the interplay between modulus, fracture resistance, and lubricity. Enhancing one property may diminish another, making this a significant challenge for material scientists and engineers.
Conclusion
The “psychorheological” experience of cutting tissue is shaped by a combination of mechanical, rheological, and structural factors. As the field advances, understanding and measuring these properties—modulus, fracture resistance, and lubricity—will be crucial for developing realistic, reliable, and ethical alternatives for surgical training and device development.
At Cambridge Polymer Group, we are committed to advancing the science of tissue simulation, helping to bridge the gap between synthetic models and the real-world demands of medical practice. If you’d like to learn more about how we can support your projects, please reach out!
Posted by Catherine Cerasuolo on | Comments Off on Squeezing the Most Out of Medical Device Hydrogels Webinar
Wednesday, August 13, 2 p.m. EDT
Hydrogels are rapidly transforming the medical device landscape, offering material properties that more closely emulate natural tissues than traditional rigid alternatives. In the upcoming webinar, “Squeezing the Most Out of Hydrogel Medical Devices,” Dr. Gavin Braithwaite will provide an in-depth perspective on how these unique polymers are advancing the field, what considerations must be made when designing with them, and how both hydrogel testing and regulatory pathways are struggling to keep pace.
What Makes Hydrogels Special in Medicine?
Hydrogels, found naturally in places like the vitreous of the eye and cartilage of the knee, are networks of polymers that retain large amounts of water, combining the flexibility of liquids with the structural integrity of solids. This duality makes them especially suited for medical device applications where a material needs to interact harmoniously with human tissue—for example, soft contact lenses, wound dressings, and implantable devices. The morphology of hydrogels and their dynamic response to environmental conditions encourages their use in combination products where both therapeutic drug release and mechanical properties are needed.
Selecting and Designing with Hydrogels
Dr. Braithwaite will detail the complex process of choosing the right hydrogel for specific device needs. Unlike metals or plastics, hydrogels offer a vast chemical and physical palette, allowing engineers to tune stiffness and damping, porosity, solutes and water content to suit a particular biological function. These tunable hydrogel properties require designers to weigh numerous factors, including chemistry, polymer structure, temperature response, network form, and the end-use environment, to ensure the device performs as intended over its lifecycle. This customization means designers must weigh numerous factors:
Hydrogel chemistry: The base polymers selected greatly impact biocompatibility and durability. Intentional degradation behavior can be designed into the chemistry.
Structure and architecture: Network density and cross-linking affect performance and response in the body.
End-use environment: Considerations like exposure to fluids, enzymes, or physical stress guide design choices.
Testing Challenges: Not Just Any Protocol Will Do
One of the standout points in the webinar will be the unique testing and characterization challenges posed by hydrogels. Standard tests designed for hard plastics or metals often fall short when used on soft, dynamic materials like hydrogels. Dr. Braithwaite will highlight several critical areas:
Fatigue testing: Hydrogels experience wear in very different ways than rigid materials.
Thermal aging: Their water-rich nature means temperature changes can alter properties considerably.
Biocompatibility: Absorption of aqueous solvents and expulsion of water from the hydrogel in non-polar solvents can make assessment of biological safety challenging.
Testing protocols must be adapted or reinvented to accurately assess the safety and longevity of hydrogel devices.
The Regulatory Maze for Hydrogel Devices
The regulatory landscape is another domain where hydrogels face unique obstacles. Many established standards were originally developed for rigid materials and can present mismatched requirements for hydrogels. Dr. Braithwaite will explain:
Legacy tests may not be “fit for purpose” for soft materials.
Evolving standards: Developers sometimes need to work with regulators to establish new or modified test methods for hydrogels.
Impact on innovation: These regulatory complexities can slow development and approval of innovative hydrogel-based devices.
Moving Forward: Balancing Opportunity and Challenge
Dr. Braithwaite’s session will emphasize both the potential of hydrogels, from mimicking real tissue to enabling next-generation therapies, and the hurdles that still slow their adoption. From the chemistry bench to regulatory filings, every step demands careful consideration and sometimes, entirely new approaches.
Key Takeaways:
Hydrogels are reshaping how we replicate and repair human tissue in medicine.
Material selection requires a careful balance of chemistry, structure, and intended use.
Standard testing and regulations often need significant adaptation for these polymers.
Close collaboration with regulatory bodies is crucial for successful device approval.
Although this webinar took place on Wednesday, August 13, 2025, the recording is now available. For anyone interested in the intersection of materials science and medical innovation, this webinar is your opportunity to learn how hydrogels are shaping the future of medical devices and what it takes to bring new hydrogel-based solutions to market. View the recording today!
Posted by Dr. Stephen Spiegelberg on | Comments Off on Flamingos Doing Vector Calculus
While we’ve previously celebrated the kitschy charm of plastic flamingos, today we turn our attention to the remarkable living birds and the science behind their mesmerizing feeding behaviors. With their beaks and most of their heads submerged near their feet, the birds stomp their feet in a rhythmic manner while chattering their beaks. But what exactly are they doing beneath the surface?
Unlocking the Flamingo’s Secret Techniques
Driven by curiosity, Víctor Ortega-Jiménez from the University of Maine, alongside collaborators from Georgia Tech and Kennesaw State University, decided to find out. Using detailed, 3D-printed models of flamingo heads, the team recreated and analyzed the birds’ signature feeding actions. Their findings, published in the Proceedings of the National Academy of Sciences (PNAS) in May 2025, finally unveiled the sophisticated strategies flamingos use to feed—movements that have captivated both bird enthusiasts and scientists for years.
The Dance That Drives Dinner
Flamingos perform a sort of underwater ballet: spinning and stomping their webbed feet in circles, stirring up the muck below. Far from random, this “wading dance” is a carefully choreographed routine designed to conjure swirling currents—vortices—that lift shrimp and other tiny morsels from the lakebed. The circular motion funnels these snacks into the water column, right where the flamingo’s beak can reach them.
Beak Work: Precision and Power
With their heads submerged, flamingos rapidly chatter their beaks up to a dozen times per second while their tongues pulse in sync. This rapid-fire action generates suction and whirlpools, channeling food particles toward the beak’s tip. As flamingos sweep their beaks backwards, these miniature vortices gather prey, making each mouthful more efficient.
The Grand Finale: The Head Lift
Every so often, flamingos abruptly lift their heads, creating a final swirl of water that draws even more food upward. This dynamic combination of footwork, beak action, and sudden head movements transforms the flamingo into an active predator, not just a passive filter feeder. Every part of their anatomy—from flexible feet to uniquely shaped beaks—works in concert to manipulate water and maximize their feeding success. These birds have studied and mastered complex chaotic fluid mechanic predictions, something that engineers sweat in their final years of undergraduate degrees.
Inspiration for Technology
The lessons learned from flamingo feeding could spark innovations in water filtration, microplastic collection, and aquatic robotics. By mimicking how flamingos harness fluid dynamics, engineers might develop new ways to capture tiny particles from water, offering nature-inspired solutions to modern challenges.
See Flamingos in Action
For those eager to witness these pink mathematicians at work, check out the videos and supplementary materials in the PNAS journal article.
Posted by Cambridge Polymer Group on | Comments Off on Balancing Analytical Uncertainty and Toxicological Risk Assessment
Tentative Identifications in Medical Device Chemical Characterization
Analytical chemical characterization and toxicological risk assessment are essential for evaluating the risk posed by chemicals that may be present in medical devices. As detailed by ISO10993-17 and ISO 10993-18, this process can support the biological safety of a device through assessment of the toxicological risk of extractables and leachables.
The Role of Mass Spectral Expertise in Identification
In practice, the burden of identification of these chemicals relies heavily on mass spectrometry and the expertise of analytical chemists, without guidance from toxicologists. Confidence levels are assigned to each analyte based on the quality of the spectral data and the availability of reference standards. These confidence levels range from confirmed identifications to unknown, along with tentative or partial assignments, reflecting the real-world complexity of chemical analysis.
The Reality of Tentative Identifications
As highlighted in the recent article, “Unknown Confidence in Chemical Characterization Identification Levels: When Tentative Identifications Are Adequate for Toxicological Risk Assessment of Medical Devices,” are common: a review of approximately 600 chemical characterization reports from a range of laboratories, including manufacturer-operated analytical facilities and contract research organizations, found that about 43% of reported organic compounds were only tentatively identified. Although there has been a recent push for confirmed or confident identifications for toxicological risk assessment, ISO 10993-17 does not specify how to handle varying confidence levels. As such, through collaboration between toxicologists and chemists, a pragmatic approach that balances analytical rigor with practical constraints is possible.
Rather than evaluating every compound independently, the article highlights grouping chemicals with similar structures for toxicological risk assessment. This approach allows for efficient evaluation of potential hazards, even when full identification is not possible. To support this method, the authors developed a decision tree that encourages early communication between chemists and toxicologists and helps analysts determine when additional analytical information is needed to improve compound identification. This structured process ensures that the toxicological risk posed by all chemicals can be assessed appropriately, including those of higher risk that may require a more detailed investigation.
The article’s insights are informed by the expertise of co-author Rebecca (Becky) Bader, PhD, Director of Regulatory Services at Cambridge Polymer Group. With over 20 years of experience in polymeric materials, drug delivery, and analytical chemistry, Becky brings deep expertise from both industry and academia. Her leadership at CPG is instrumental in advancing analytical techniques and ensuring that chemical characterization studies meet the highest standards of scientific rigor and regulatory compliance.
Join Our Upcoming Webinar The Tightrope of Tentative IDs: Balancing Analytical Uncertainty with Toxicological Risk Assessment
July 9, 2025 | 2:00 PM EDT
Don’t miss this opportunity to dive deeper into balancing analytical uncertainty and toxicological risk! Study authors Becky Bader and Steph Street, a Senior Principal Toxicologist and Biocompatibility SME for Medtronic, will host a webinar discussing:
Regulatory expectations regarding compound identification and toxicological risk assessment
The grouping of chemical compounds, including those with tentative identifications, for toxicological risk assessment
Strategies for streaming lining the chemical characterization and toxicological risk assessment process
Case studies on collaborative chemist-toxicologist workflows
Cambridge Polymer Group specializes in comprehensive material and chemical characterization services, including extractables and leachables testing for medical devices. Our team, led by experts like Becky, leverages analytical instrumentation and deep knowledge of polymer science to:
Design and execute tailored extractables and leachables studies
Identification of unknowns for toxicological risk assessment
Provide clear, defensible reports for regulatory submission
Guide clients through the decision-making process, from data collection to risk assessment and regulatory strategy
Every medical device is unique, and our approach ensures that analytical methods and risk assessments are customized to your product’s specific materials, manufacturing processes, and intended use. Whether you need targeted testing or a comprehensive chemical risk assessment, CPG’s experience and expertise can help you navigate the evolving regulatory landscape and bring safe, effective devices to market.
Posted by Dr. Stephen Spiegelberg on | Comments Off on Potential Changes to the Generally Recognized as Safe (GRAS) Program
Background: The GRAS Framework
The Food Additives Amendment to the Federal Food, Drug, and Cosmetic Act (FD&C Act) was established by Congress in 1958. In the Code of Federal Regulations, the rules that the FDA applies to food additives are spelled out in sections 21 CFR 170.3 and 170.30. A food additive is considered to be any substance that is intentionally added to food or may reasonably be expected to become a component of food, such as leachable components from packaging. These additives are required to be reviewed and approved by the FDA before the additives can be used in food products as part of a premarket approval process.
However, there are exceptions to this review requirement. If the substance is Generally Recognized by qualified experts As having been adequately shown to be Safe (GRAS) under the conditions of its intended use, the substance does not require FDA approval and is not considered a food additive. GRAS assessment can be performed through scientific analysis, or from safe historical consumption of the substance if it has been used in food prior to 1958.
The Self-Affirmation Pathway and Its Controversy
Since 2016, the FDA has operated a voluntary GRAS notification program. Under this system, any qualified individual can notify the FDA that a substance is not subject to the premarket approval process as it is considered GRAS. The FDA may not question the basis for the GRAS conclusion, or it may conclude that there is insufficient information to make a GRAS conclusion.
Although the FDA had a GRAS affirmation process in place around 1972, it was discontinued by 1997 due to lack of resources and was replaced with the notification process. The FDA maintains a GRAS database of notifications. The GRAS list, which is not comprehensive, is located in 21 CFR 182, 184, and 186.[1] Notably, the GRAS notification process is voluntary, and does not require either notification or affirmation from the FDA.
This self-affirmation pathway has been criticized as a “loophole,” enabling manufacturers to introduce new food ingredients without sufficient safety data or transparency. While the process allows for efficiency and rapid market entry, it also means that the FDA and consumers may be unaware of new substances in the food supply.
Proposed Changes in 2025
In March 2025, the Health and Human Services secretary directed the FDA to consider removing the self-affirmation process of the GRAS program.[2] Companies would need to publicly notify the FDA of their intended use of substances in food products, along with safety data, before they could go to market with the substances. This substantial change in legislation would require many companies to re-evaluate their safety data and may require retroactive approval from the FDA.
Current vs. Proposed GRAS Process
Aspect
Current GRAS Program
Proposed Changes (2025)
FDA Notification
Voluntary
Mandatory
Public Disclosure
Not required
Required
FDA Premarket Review
Not required
Required
Industry Burden
Lower
Higher
Transparency
Limited
Enhanced
Time to Market
Shorter
Longer
Implementing these changes will not be immediate. The FDA must conduct formal rulemaking, and because the GRAS exemption is written into federal law, Congressional action may be required. These steps could take years and may face industry resistance and legal challenges.
Establishing Safety Profiles for Food Additives
Deliberately Added Ingredients: Toxicological evaluation of the ingredients based on the chemistry and amount can assist in establishing the safety profile.
Inadvertent Additives (e.g., from Packaging): Inadvertent food additives may be introduced from materials contacting food products, including food processing equipment, containers, or food preparation surfaces. In these cases, substances may diffuse into the food from the contact materials, which are often plastic and may contain antioxidants, colorants, plasticizers, and other stabilizers. For these substances, migration testing needs to be performed using food simulants to assess the amount of substance that is anticipated to be incorporated into the food product. This testing is comparable to leachables/extraction testing performed for medical devices.
Posted by Cambridge Polymer Group on | Comments Off on ASTM Workshop on the Characterization of Hydrogel Medical Devices: Key Takeaways
On May 6, 2025, the ASTM Workshop on the Characterization of Hydrogel Medical Devices brought together researchers and engineers to discuss current test methods for hydrogels in medical devices. Led by Stephen Spiegelberg of Cambridge Polymer Group, the workshop focused on current test methods, industry challenges, and the need for new standards.
Why Are ASTM Standards and Workshops Important?
ASTM standards play a crucial role in the medical device industry by:
Establishing best practices for testing methods for researchers, especially those new to the field.
Improving repeatability and accuracy across different laboratories.
Assisting regulatory agencies in verifying the quality and reliability of submitted data.
Providing companies with confidence that their test methods will withstand regulatory scrutiny.
ASTM workshops are designed to:
Share the latest understanding and best practices on the topic area within the industry.
Gather feedback from regulators on test methods to facilitate regulatory clearance.
Identify gaps in current testing methods and associated standards.
Establish task groups to develop new and improved standards.
Identifying Hydrogel Gaps and Needs in Hydrogel Characterization
A notable finding of the May 6th workshop was that only two relevant standards for hydrogel testing currently exist across ASTM, ISO, and USP. This lack of established guidance highlights a significant unmet need, especially as hydrogels are being used more often as structural components rather than just as coatings.
Development of animal models for safety and effectiveness testing
Evaluation of high-water-content hydrogels
Characterization of degradable and specialized hydrogels
Standardization Priorities
During a closing discussion led by co-chair Jon Moseley, participants identified several top priorities for new standards, with the development of a common terminology for hydrogels emerging as a particularly urgent need. Inconsistent language can create confusion among manufacturers, regulators, and end users, so establishing clear definitions is essential.
Other priorities for standardization include:
Friction measurements
Mechanical testing methods
Dynamic property assessment (rheology and DMA)
Accelerated aging protocols
Environmental conditioning
Chemical risk assessment, particularly regarding solvent selection
Mechanical testing and accelerated aging generated the most discussion, as they appear to be the most challenging currently. Chemical risk assessment was also a discussion, particularly with regards to solvent selection for chemical characterization. Task groups are being formed to address these topics, and participation from those with relevant experience is encouraged.
Looking Forward: Opportunities and Advice
For those new to hydrogels, it’s important to recognize that standard test methods for other polymers, such as thermoplastics, elastomers, and thermosets, may not be suitable due to hydrogels’ unique properties and greater batch-to-batch variability. As one participant aptly summarized,
“Hydrogels always find a way to mess with you.”
Manufacturing hydrogel devices presents ongoing challenges related to their compliance, temporal variability, and unique chemistries. As hydrogels are used in more advanced applications, such as degradable implants or piezoelectric devices, the need for robust, widely accepted testing standards will only grow. Regulatory requirements are currently quite stringent for hydrogels, particularly degradable ones, due in large part to lack of industry-wide experience with these materials.
Collaboration between experienced developers and regulatory agencies will be vital as new standards are developed. If you are interested in contributing to these efforts, please contact Cambridge Polymer Group at info@campoly.com. Stay tuned for further updates as the ASTM task groups work to advance hydrogel testing standards and support innovation in medical device development.
 Dr. Kalpana Viswanathan is a seasoned polymer chemist with over ten years of research and development experience. Her portfolio includes pioneering coatings for implantable medical devices and
Dr. Kalpana Viswanathan is a seasoned polymer chemist with over ten years of research and development experience. Her portfolio includes pioneering coatings for implantable medical devices and